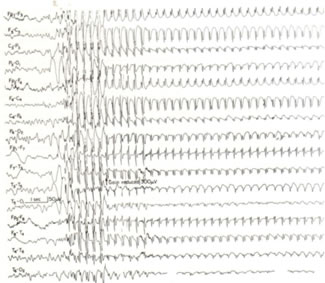
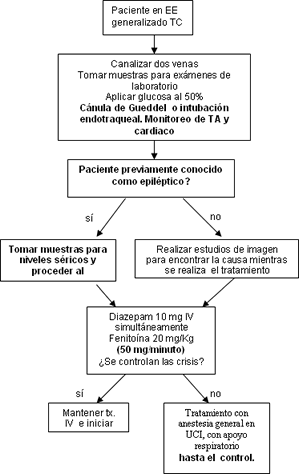
El tratamiento de la epilepsia
con medicamentos se inició el
siglo XIX con la utilización de los bromuros. Desde entonces,
se han desarrollado diversos fármacos que son actualmente de
uso común, y se encuentran otros más en investigación.
Cualquiera que sea el medicamento, necesariamente lleva a cabo diversos
pasos desde su ingestión hasta la producción de efectos
clínicos
Los cambios que los medicamentos
(fármacos o drogas) sufren
en el organismo se resumen en el cuadro siguiente:
|
DROGA
|
ABSORCION
%
|
UNION
A PROTEINAS HS.
|
VIDA
MEDIA
|
RUTA
DE ELIMINACION
|
CARACTERISTICAS
ESPECIALES
|
|
FENOBARBITAL
|
Lenta(95-100%)
|
48-54
|
72-144
|
Metab
hep. 25% sin cambios
|
Inductor
enzimático, Sedante
|
|
FENITOINA
|
Lenta
(85-95%)
|
90-93
|
9-40
|
Metab.hep.
saturable Vida media dependiente de
de la concentración
|
Inductor
enzimático
|
|
PRIMIDONA
|
Rápida
(90-100%)
|
20-30
|
4-12
|
Metab.hep.Metabolitos
excretados sin cambios (40%)
|
Inductor
enzimático
|
|
CARBAMAZEPINA
|
Lenta
(75-85%)
|
70-78
|
8-24
|
Metab.hep.
Metabolito activo: Autoinduce su metabolismo
|
Inductor
enzimático
|
|
ACIDO
VALPROICO
|
Rápida
(100%)
|
88-92
|
7-17
|
Met.hep.
Metabolitos activos Concentración dependiente de unión
a proteínas
|
Inhibidor
enzimático
|
|
CLONAZEPAN
|
Rápida
(80-90%)
|
80-90
|
30-40
|
Met.hep.Produce
tolerancia
Exacerbación al suspenderlo
|
Sedante
|
Al igual que cualquier medicamento,
los fármacos anticonvulsivos
tienen efectos colaterales que en ocasiones son severos y pueden obligar
a descontinuar el tratamiento.
Los medicamentos anticonvulsivos
interactúan entre sí,
por lo que lo más recomendable es evitar asociaciones. Ocasionalmente éstas
pueden intentarse y en esos casos es necesario tener en mente los efectos
recíprocos sobre los respectivos niveles:
|
|
DFH
|
CBZ
|
FB
|
PMD
|
VPA
|
BZD
|
|
DFH
|
.
|
.
|
.
|
+
|
-
|
.
|
|
CBZ
|
-
|
.
|
.
|
+
|
-
|
-
|
|
FB
|
.
|
-
|
.
|
.
|
-
|
.
|
|
PMD
|
.
|
-
|
.
|
.
|
.
|
.
|
|
VPA
|
-
|
+
|
+
|
+
|
.
|
.
|
|
BZD
|
+
|
.
|
.
|
.
|
.
|
.
|
Con los fármacos anticonvulsivos conocidos es posible controlar
satisfactoriamente más del 80 % de los casos de epilepsia, aunque
no siempre con el primer tratamiento elegido.
Como mencionamos en los lineamientos generales, lo más recomendable
es la monoterapia (terapia con un solo medicamento). pues con las asociaciones
de medicamentos es difícil saber a cuál droga atribuir tanto
los efectos terapéuticos como los indeseables y éstos aumentan
con las interacciones.
La cuantificación de los niveles séricos de los fármacos
anticonvulsivos es un auxiliar valioso. Al inicio de un tratamiento sirven
como una guía para ajustar dosis siempre y cuando se realicen pasado
el tiempo necesario para alcanzar el estado estable (cuando los niveles del
fármaco circulante tienen mínimas fluctuaciones). Si el paciente
se controla bien y no hay manifestaciones colaterales es innecesario repetir
los niveles periódicamente, pero si no hay buen control o hay efectos
colaterales o tóxicos es conveniente hacerlos para decidir las modificaciones
pertinentes.
FÁRMACOS TRADICIONALES
Los llamados fármacos tradicionales son aquéllos que
tienen en uso varias décadas y cuyos efectos tanto terapéuticos
como colaterales y tóxicos son ya muy bien conocidos: fenobarbital,
fenitoína, primidona, carbamazepina, valproatos y benzodiazepinas,
que se describirán en sus mecanismos de acción, indicaciones,
efectos colaterales y tóxicos.
Fenobarbital. S e introdujo a
la clínica en
1912 como hipnótico y tranquilizante, pero se descubrió por
Hauptmann ese mismo año que las crisis epilépticas tenían
una marcada reducción con su uso. .
Indicaciones: crisis tónico-clónicas
generalizadas y crisis parciales.
Efectos colaterales y tóxicos: sedación, ataxia, mareo,
insomnio, hiperquinesia, cambios del talante, agresividad, disfunción
cognitiva, impotencia, reducción de la libido, deficiencia de
folato, de vitamina K y D, osteomalacia, contractura de Dupuytren,
hombro congelado, anormalidades del tejido conectivo, rash.
A pesar de los efectos colaterales, es muy efectivo
y barato. Muchos de los efectos son dependientes de dosis, por lo
que una vigilancia adecuada, con la dosis mínima efectiva
mejoran la tolerabilidad.
Fenitoína. (Wilder) Merritt
y Putnam en 1938 descubrieron el efecto anticonvulsivo de la fenitoina
sin efecto hipnótico,
lo cual significó un cambio muy notable en el tratamiento de
la epilepsia, hasta entonces a base de bromuros y fenobarbital y fue
el primer producto que mostró efectividad anticonvulsiva en
animales antes de ser probado en seres humanos.
Indicaciones: crisis parciales y crisis generalizadas
tónico-clónicas.
Efectos colaterales y tóxicos: ataxia, mareo, letargia, sedación,
cefalea, disquinesia,, encefalopatía aguda, cambios de talante,
depresión, agresión, síndrome cerebeloso crónico,
neuropatía periférica; datos de hipersensibilidad (erupción
cutánea, fiebre, anemia); deficiencia de folatos, anemia megaloblástica,
afectación de la médula ósea; hepatitis, deficiencia
de vitamina K, defectos de coagulación; disminución de
las inmunoglobulinas (proteínas que constgituyen los anticuerpos
o defensas del organismo), descalcificación, disfunción
tiroidea, disfunción hormonal, pérdida de la libido,
trastornos del tejido conectivo, pseudolinfoma, vasculitis, miopatía;
rasgos faciales toscos, hirsutismo (exceso de veloo en las mujeres),
hinchazón de las encías.
Carbamazepina. . A finales de
los años 50
se probó exitosamente como anticonvulsivo y como tratamiento
de la neuralgia del trigémino, estableciéndose en los
siguientes años como uno de los anticonvulsivos más importantes
del mundo.
Indicaciones: crisis parciales y crisis generalizadas
tónico-clónicas.
Efectos colaterales y tóxicos: somnolencia, fatiga, mareo,
ataxia, diplopia, visión borrosa, rash y otras reacciones cutáneas,
discrasias de médula ósea, hiponatremia, retención
de agua y nefritis.
Valproato. B.S. Baron lo sistetizó por pirimera
vez en 1882 y fue usado inicialmente como un solvente orgánico.
Meunier, en 1963, fue el primero que identificó au acción
antiepiléptica mientras lo usaba como solvente en en el tamizaje
de nuevas drogas antiepilépticas. Los primeros estudios clínicos
fueron reportados por Carraz en 1964 y desde entonces su uso ha sido
cada vez más difundido.. Del ácido valproico se han derivado
el valproato de magnesio, de sodio y el divalproato de sodio.
Indicaciones. Crisis parciales y todo tipo de crisis
generalizadas (tónico-clónicas, ausencias, mioclonias, atónicas).
Efectos colaterales y tóxicos: Toxicidad hepática severa
en niños pequeños (efecto raro), pancreatitis, insuficiencia
renal, mareo, trastornos cognitivos, agresividad, temblor, debilidad,
encefalopatía, trombocitopenia, neutropenia, anemia aplástica,
adelgazamiento y pérdida del cabello, ganancia de peso .
Existen nuevos fármacos (oxcarbazepina, lamotrigina, felbamato, gabapentín,
vigabatrina, topiramato, levetiracetam, etc.) que actualmente se usan en los
casos considerados refractarios y cuya verdadera utilidad se evaluará con
el tiempo.
Fármacos tradicionales
Nombre genérico |
Abreviatrura |
Marcas disponibles |
| Fenobarbital |
Pb |
Fennabbott |
| Fenitoína |
PHT |
Epamín, Fenidantoín S, Fenitrón |
| Primidona |
PMD |
Mysoline, Pridona |
| Etosuximida |
ETX |
Zarontín |
| Carbamazepina |
CBZ |
Tegretol, Neugerón, Carbazina |
| Valproato |
VPA |
Depakene (ácido valproico), Atemperator,
Criam (valproato de magnesio), Epival (divalproato de
sodio), Leptilán (valproato de sodio)
|
| Clonazepam |
CNZ |
Rivotril |
Nuevos fármacos
Nombre genérico |
Abreviatrura |
Marcas |
| Oxcarbazepina |
OXC |
Trileptal |
| Felbamato |
FBM |
Felbatol |
| Vigabatrina |
VGB |
Sabril |
| Lamotrigina |
LMG |
Lamictal |
| Gabapentina |
GBP |
Neurontín |
| Topiramato |
TPM |
Topamax |
| Levetiracetam |
LVT |
Keppra |
INDICACIONES DE LOS FÁRMACOS ANTIEPILÉPTICOS
TIPO DE CRISIS |
1ª. ELECCIÓN |
ALTERNATIVA |
| Crisis focales simples o complejas |
PHT, CBZ, VPA |
Pb, PMD, nuevos |
| Ausencias |
VPA, ETX |
LMG |
| Crisis generalizadas tónicas, clónicas y tónicoclonicas |
PHT, CBZ, VPA |
Pb,PMD, nuevos |
| Mioclónicas |
VPA, CBZ |
LMG |
| Atónicas |
CNZ, VPA |
TPM, LMG |
FÁRMACOS NUEVOS DISPONIBLES EN MÉXICO
Todos tienen en común que son producto de investigaciones encaminadas
a encontrar tratamientos antiepilépticos, han sido ensayados
en las diferentes fases de investigación y finalmente en seres
humanos antes de su comercialización. Las indicaciones iniciales
para los primeros ensayos clínicos son crisis parciales simples
o complejas con o sin generalización secundaria de difícil
control, en pacientes mayores de 14 años.. Para su evaluación
de eficacia y tolerabilidad se han comparado con carbamazepina o fenitoína
y se considera como medida de eficacia la reducción de crisis
igual o mayor al 50% en relación a la frecuencia basal, todos
utilizados como terapia de adición al esquema que previamente
tenía el paciente. La mayoría han sido ensayados ya en
niños y en síndromes epilépticos como el Lennox-Gastaut,
encontrándose otras indicaciones para algunos de ellos. Igualmente
se han ensayado como tratamiento único en epilepsia de reciente
inicio.
Oxcarbazepina. Es tan efectiva
como la carbamazepina pero es mejor tolerada y la frecuencia de efectos
indeseables, similares a los de la carbamazepina, es menor, sobre
todo en cuanto a las reacciones cutáneas.
Felbamato . Fue sintetizado por los laboratorios
Wallace en 1954.
Indicaciones: Terapia coadyuvante en epilepsia
parcial y generalizada refractarias y en el síndrome de Lennox-Gastaut.
Efectos colaterales y tóxicos: Trastorno severo del hígado
y anemia aplástica (daño en la médula ósea,
donde se producen todos los elementos de la sangre) son los más
serios, aunque raros. Otros son: insomnio, pérdida de peso,
síntomas gastrointestinales, fatiga, mareo, letargia, cambios
de conducta, ataxia, trastornos visuales, cambios del estado de ánimo,
reacción psicótica y erupciones en la piel. Se ha retirado
la aprobación de la Food and Drug Adminstration (FDA) de EUA
para su uso, debido a los efectos adversos graves.
Lamotrigina . En vista de que
varios de los anticonvulsivos tradicionales tienen efectos antifolato
(reducen una vitamina llamada ácido
fólico), se consideró que esto era una propiedad anticonvulsiva
y la investigación en ese sentido por parte de laboratorios
Wellcome dio como resultado la producción de feniltriazinas,
de las que derivó la lamotrigina, una triazina de estructura
química diferente a la de otros anticonvulsivos. Su acción
antifolato es muy débil, pero su efecto anticonvulsivo es muy
bueno, por lo que ambos efectos se consideran independientes en la
actualidad.. .
Efectos colaterales y tóxicos: erupción cutánea , dolor
de cabeza, anemia, ataxia, astenia, diplopia, náusea, vómito,
mareo, somnolencia, insomnio, depresión, psicosis y aumento
de crisis.
Vigabatrina. El GABA (Ácido Gama Amino Butírico)
es el neurotransmisor inhibidor más importante del sistema nervioso
central y sus anomalías se relacionaron con la presentación
de epilepsia, motivo por el que muchas investigaciones se encaminaron
por este rumbo. De estas investigaciones se originó la vigabatrina,
que es un análogo del GABA.
Indicaciones: Terapia coadyuvante en la epilepsia
parcial y generalizada refractarias, así como en el síndrome de West y el síndrome
de Lennox-Gastaut.
Efectos colaterales y tóxicos: sedación, mareo, cefalea,
ataxia (pérdida del equilibrio), parestesias, agitación,
amnesia, cambios del talante, depresión, psicosis, confusión,
agresividad, ganancia de peso, diarrea, trastornos del campo visual.
Gabapentina . Mecanismos de acción.
No se conocen.
Indicaciones: Terapia coadyuvante en epilepsias parcial y generalizada
refractarias.
Efectos indeseables y tóxicos: somnolencia, mareo, ataxia,
somnolencia, cefalea, temblor diplopia, náusea, vómito,
rinitis y exacerbación de crisis.
Topiramato. Tiene varios mecanismos
de acción
Efectos colaterales y tóxicos: mareo, ataxia, cefalea, parestesias,
temblor, somnolencia, trastornos cognitivos, confusión, agitación,
amnesia, depresión, labilidad emocional, náusea, diarrea
y pérdida de peso.
Levetiracetam Tiene
una absorción rápida
y casi completa, su vida media es de 6 a 8 horas. Es efectivo para
crisis parciales con o sin generalización secundaria y en epilepsia
de Janz. Se utiliza a dosis de 500 a 1500 m con buena tolerabilidad
y eficacia.
Efectos adversos:
somnolencia, astenia, infección, mareo, alteraciones
psiquiátricas.
Uso de la dieta catogénica en epilepsia
infantil
Por: Dra. Eneida Porras Kattz
La dieta cetogénica clásica se desarrolló en
1920 para el tratamiento de niños con crisis de muy difícil
control, y en la actualidad continúa su uso en pacientes que
han tenido mala respuesta a los medicamentos antiepilépticos.
Consiste en una alimentación alta en grasas y baja en carbohidratos
y proteínas, la cual produce los llamados cuerpos cetónicos derivados
de la digestión de las grasas, Estos cuerpos cetónicos aumentan
la acidez de la sangre y contribuyen a mejorar el control de cualquier tipo
de crisis. Las que mejor responden son las crisis mioclónicas (sacudidas
o sobresaltos repentinos) y las atónicas (caídas súbitas
al aflojarse el cuerpo, sin que el paciente se dé cuenta de ello).
La alimentación se calcula en forma muy cuidadosa de acuerdo a la edad,
el peso y la estatura del paciente, tomando en cuenta sus necesidades calóricas
de crecimiento.
Aunque se desconoce el mecanismo exacto por el cual actúa la dieta cetogénica,
se ha postulado que influye en forma favorable en el metabolismo energético
cerebral, e incrementa las reservas energéticas cerebrales.
La dieta cetogénica se utiliza como un último recurso, cuando
las crisis producen incapacidad a pesar del uso combinado de dos o tres medicamentos
o cuando el medicamento ha tenido los reajustes necesarios por un período
de meses o años, sin éxito alguno. Se prescribe en niños
entre 1 a 8 años de edad; los niños más pequeños
tienen dificultad para alcanzar el estado cetósico y hay tendencia a
la hipoglicemia (niveles de azúcar en sangre por debajo de lo normal);
por arriba de esta edad es más difícil que cumplan con la dieta.
Si bien es generalmente bien tolerada, no está libre de efectos colaterales
potenciales, entre ellos, reducción significativa de la masa ósea
(disminución del calcio depositado en los huesos), formación
de cálculos renales, adelgazamiento del cabello y caída del mismo,
complicaciones cardiovasculares asociadas a aterosclerosis (depósito
de grasa en las paredes de las arterias), así como alteraciones en el
estado de conciencia, que pueden llegar hasta el estado de coma.
El inicio de la dieta requiere de que el paciente se interne en un hospital
con la finalidad de enseñarle a él y a su familia cómo
llevarla a cabo, vigilar el estado metabólico del paciente y realizar
un registro inicial sobre la respuesta del paciente a la dieta en cuanto
al control de las crisis.
Los resultados con la dieta cetogénica son variables, pero se reporta
hasta el 40% de reducción de las crisis en más de un 50% de los
casos.
El éxito de la dieta cetogénica depende de varios factores, pero
primordialmente es el resultado de la participación de un equipo médico
multidisciplinario, de un apoyo familiar muy amplio y de una gran motivación
por parte del paciente y la familia para iniciar y mantener el rigor que la
dieta exige.
TRATAMIENTO ANTIEPILÉPTICO DURANTE
EL EMBARAZO .
El uso de medicamentos durante el embarazo incrementa el riesgo de
malformaciones, sin embargo, el descontrol de la epilepsia resulta
más peligroso, así como la polifarmacia (uso de muchos
medicamentos al mismo tiempo). Por lo tanto debe usarse un solo medicamento,
y el de elección es el que controla mejor a la paciente.
TRATAMIENTO DE LA MUJER EPILÉPTICA DURANTE
EL EMBARAZO
La mujer epiléptica puede tener descendencia si así lo
desea, pues la mayoría de las epilepsias no son hereditarias
.pero el uso de medicamentos durante el embarazo incrementa el riesgo
de malformaciones en relación a la población general
(en la población no epiléptica es del 2% y con tratamiento
antiepiléptico aumenta al 6%), sin embargo, el descontrol de
la epilepsia resulta más peligroso, así como la polifarmacia
(uso de muchos medicamentos al mismo tiempo). Por lo tanto debe usarse
un solo medicamento, y el de elección es el que controla mejor
a la paciente .
Es aconsejable que la mujer comunique
al neurólogo tratante
la decisión de embarazarse desde que es una intención,
pues desde antes de la concepción debe disminuirse el riesgo
de teratogénesis administrando ácido fólico, vitamina
que se sabe se reduce con el uso de los fármacos antiepilépticos
y cuya carencia se relaciona con la presentación de malformaciones
congénitas. Actualmente, muchos neurólogos deciden administrarla
a toda mujer que ha empezado a reglar, puesto que los embarazos inesperados
son frecuentes y la joven con epilepsia debe estar informada de antemano
de los riesgos reales y cómo prevenirlos. Durante el embarazo,
la epilepsia puede mantener un control similar al que tenía
previamente, mejorar o empeorar, y el mejor tratamiento antiepiléptico
para la mujer embarazada es el que mejor la controla. Lo más
deseable es que el control de las crisis sea muy estricto, ya que su
presentación frecuente puede provocar incluso pérdida
del producto o al menos daño cerebral. Por lo tanto, la vigilancia
debe ser mayor, tanto desde el punto de vista neurológico para
el control de la epilepsia como obstétrico, para conocer el
desarrollo del bebé, posición, etc. . Las decisiones
en relación al tratamiento antiepiléptico competen exclusivamente
al neurólogo tratante, quien deberá auxiliarse más
frecuentemente de los niveles séricos y ajustar periódicamente
las dosis para mantenerlos en rangos terapéuticos con la mínima
cantidad de medicamento que se necesite. En el último mes del
embarazo puede requerirse la administración de vitamina K para
prevenir una hemorragia en el bebé, si el tratamiento es con
fenobarbital y se recomienda la aplicación de vitamina K I.M.
al final del embarazo. Si el parto será por vía natural
o por cesárea, es una decisión que tomará el ginecólogo
basándose en las condiciones de la mujer y no porque exista
epilepsia.